Featured Articles
N-glycans in protein folding: Operating unchaperoned
Functional Glycomics (08 April 2009) | doi:10.1038/fg.2009.14Standfirst
Core N-glycans of the cystic fibrosis transmembrane conductance regulator (CFTR) enhance its folding and stability, independently of lectin-like chaperones
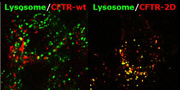
The destabilized, glycosylation-deficient CFTR (2D) is targeted into dextran-loaded lysosomes from the plasma membrane, whereas the wild-type channel avoids lysosomal delivery and recycles
N-glycosylation, recognized by endoplasmic reticulum chaperones during folding cycles, is an essential co-translational modification for the correct folding, stability and membrane expression of numerous proteins. Folding of the CFTR is complex, relies on chaperones and is easily disrupted by mutations that cause cystic fibrosis. Lukacs and colleagues have now shown in the Journal of Cell Biology that CFTR N-glycans, in addition to recruiting chaperones, also play an independent role in folding and traffic of the protein.
The modular, multi-pass transmembrane structure of the CFTR makes it difficult to assemble correctly, and only a small proportion of nascent chains are correctly folded and transported to the cell surface in coat protein complex II (COPII) vesicles. The protein is known to rely on calnexin (CNX), a lectin-like endoplasmic reticulum chaperone, which assists CFTR folding by binding the core glycan chains of folding intermediates. Two CFTR N-glycosylation sites were mutated by the authors, singly or together; their importance for stability was reflected by respective surface expression reductions of  37%, 63% and 87%. Pulse-chase experiments confirmed that folding efficiency in these mutants was comparably reduced, and that CNX was unable to bind the double mutant. However, surprisingly, the authors found that disruption of the wild-type CFTR–CNX interaction by inhibition of core glycan modification or by CNX knockdown accounted for only about 35% of the reduction in folding efficiency, compared with complete inhibition of glycosylation. Glycans therefore play a role in CFTR folding efficiency independently of their ability to bind CNX.
37%, 63% and 87%. Pulse-chase experiments confirmed that folding efficiency in these mutants was comparably reduced, and that CNX was unable to bind the double mutant. However, surprisingly, the authors found that disruption of the wild-type CFTR–CNX interaction by inhibition of core glycan modification or by CNX knockdown accounted for only about 35% of the reduction in folding efficiency, compared with complete inhibition of glycosylation. Glycans therefore play a role in CFTR folding efficiency independently of their ability to bind CNX.
Although disruption of the CFTR–CNX interaction reduced folding efficiency, it did not affect the metabolic or cell-surface turnover rate of the mature CFTR, whereas inhibition of glycosylation and the double mutation both destabilized the channel threefold. Enzymatic de-glycosylation of the mature channel did not reduce stability; glycans are essential for productive folding but are not required for maintenance of the native fold once achieved. The mutant and wild-type channels that did reach the cell surface were internalized at a comparable rate. However, monitoring the reappearance of labeled CFTR at the surface showed that recycling was reduced by about 60% by the single mutations, and by more than 80% by the double mutations. The internalized wild-type channel was directed to recycling endosomes, whereas the mutants accumulated in lysosomes and their ubiquitination level was substantially raised. The protease-resistance of the mature double mutant was reduced compared with wild type, particularly in the second membrane-spanning domain, and its propensity for thermoaggregation was increased.
This study illuminates the complex role of N-glycans in CFTR biogenesis. It also emphasizes the impact of folding energetics on membrane protein turnover at both the early secretory pathway and the later endosomal system. Conformational destabilization caused by the absence of N-glycans correlates with ubiquitination and lysosomal targeting, representing a quality control mechanism for integral membrane proteins in post-Golgi compartments. This may serve as a model for currently unexplained raised turnover rates of other glycosylation-deficient proteins at the plasma membrane.