Featured Articles
N-linked glycosylation: A reaction takes shape
Functional Glycomics (07 July 2011) | doi:10.1038/fg.2011.25Standfirst
A 3.4-Å structure of the Campylobacter lari oligosaccharyltransferase provides the first detailed view of protein structure, substrate binding and asparagine activation in the most common modification reaction in glycobiology.
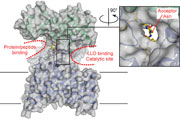
Image courtesy of Kaspar Locher.
N-linked glycosylation involves the transfer of a carbohydrate group from a lipid-linked oligosaccharide to the asparagine residue in an N-X-S/T motif of a substrate protein; catalysis of the reaction by an oligosaccharyltransferase (OST) is required to overcome the poor reactivity of the asparagine's amide group. OSTs can be single proteins or multiprotein complexes. Most structural work has focused on the single, bacterial enzymes, and more specifically on the periplasmic domain of these integral membrane proteins. However, the isolated periplasmic domain is catalytically inactive, so it has not been possible to understand how the enzyme orchestrates carbohydrate transfer. Kaspar Locher and colleagues, reporting in Nature, now reveal the structure of a complete OST — PglB from C. lari — that both explains the inactivity of the periplasmic domain and outlines a possible mechanistic course.
PglB was crystallized with a hexapeptide substrate mimic containing the typical N-X-T motif and an aspartic acid residue at the −2 position, known to be important for bacterial OSTs. Although the periplasmic domain looks as expected based on prior work, the extensive contacts between this domain and the transmembrane domain, which defines a novel fold, are a surprise. The authors also observed two large cavities at the interface between the protein domains, with the hexapeptide found in one of these cavities. Previous work had suggested that the +2 S/T residue from the substrate might be involved in catalysis, but the structure demonstrates that the hydroxyl group of this residue is engaged in a well-defined network of hydrogen bonds to the strictly conserved WWD motif of the enzyme. Packing of the threonine residue's methyl group against a neighboring isoleucine also demonstrates why threonine is strongly preferred over serine in the substrate motif. A salt bridge between the −2 Asp and Arg331 — a residue conserved in bacterial but not in eukaryotic OSTs — explains the basis for the extended bacterial motif.
In the second cavity, the authors discovered a metal ion coordinated to the known D-X-D motif, but also identified two additional residues — Asp56 and Glu319 — that bind to this metal and form hydrogen bonds to the substrate asparagine. The direction of these hydrogen bonds led the authors to postulate that these interactions are able to rotate the reactive nitrogen out of plane from the carbonyl group, thus restoring nucleophilicity to this atom and promoting the reaction. Accordingly, mutations to these acidic residues reduced enzyme activity significantly.
Finally, the authors create a model of the enzyme with the lipid-linked oligosaccharide bound in the catalytic site, which highlights a possible dual role of the metal ion to both orient the reactive aspartate residue and stabilize the phosphate leaving group of the lipid substrate. The authors further propose that a flexible loop, EL5, has a crucial role in releasing the product, becoming fully unfolded as a result of steric crowding by the newly formed glycoprotein, and then closing again to reconstruct the catalytic site. Overall, this structure offers a wealth of new opportunities to better define the details of carbohydrate transfer and to re-engineer these enzymes to promote alternative reactions.